Rare Disease
A rare disease is any disease that affects a small percentage of the population. Most rare diseases are genetic, and thus are present throughout the person's entire life, even if phenotypes do not immediately appear. Many rare diseases appear early in life, and about 30 percent of children with rare diseases will die before reaching their fifth birthday. With a single diagnosed patient only, ribose-5-phosphate isomerase deficiency is considered the rarest genetic disease.
Human Disease Network (HDN)
A human disease network is a network of human disorders and diseases with reference to their genetic origins or other features. More specifically, it is the map of human disease associations referring mostly to disease genes. For example, in a human disease network, two diseases are linked if they share at least one associated gene. A typical human disease network usually derives from bipartite networks which consist of both diseases and genes information. Additionally, some human disease networks use other features such as phenotypes and proteins to associate diseases.
OMIM
Online Mendelian Inheritance in Man (OMIM) is a continuously updated catalog of human genes and genetic disorders and traits, with a particular focus on the gene-phenotype relationship.
UMLS
The Unified Medical Language System (UMLS) is a compendium of many controlled vocabularies in the biomedical sciences. It provides a mapping structure among these vocabularies and thus allows one to translate among the various terminology systems; it may also be viewed as a comprehensive thesaurus and ontology of biomedical concepts. UMLS further provides facilities for natural language processing. It is intended to be used mainly by developers of systems in medical informatics.UMLS consists of Knowledge Sources (databases) and a set of software tools.The UMLS was designed and is maintained by the US National Library of Medicine, is updated quarterly and may be used for free. The project was initiated in 1986 by Donald A.B. Lindberg, M.D., then Director of the Library of Medicine.
Orphanet
Orphanet is a European website providing information about orphan drugs and rare diseases. It contains content both for physicians and for patients. Its administrative office is in Paris and its official medical journal is the Orphanet Journal of Rare Diseases published on their behalf by BioMed Central.
Disease Ontology
The Disease Ontology (DO) is a formal ontology of human disease. The Disease Ontology project is hosted at the Institute for Genome Sciences at the University of Maryland School of Medicine. The Disease Ontology project was initially developed in 2003 at Northwestern University to address the need for a purpose-built ontology that covers the full spectrum of disease concepts annotated within biomedical repositories within an ontological framework that is extensible to meet community needs. The Disease Ontology is an OBO (Open Biomedical Ontologies) Foundry ontology.
ICD-10
ICD-10 is the 10th revision of the International Statistical Classification of Diseases and Related Health Problems (ICD), a medical classification list by the World Health Organization (WHO). It contains codes for diseases, signs and phenotypes, abnormal findings, complaints, social circumstances, and external causes of injury or diseases.
MeSH
Medical Subject Headings (MeSH) is a comprehensive controlled vocabulary for the purpose of indexing journal articles and books in the life sciences; it serves as a thesaurus that facilitates searching. Created and updated by the United States National Library of Medicine (NLM), it is used by the MEDLINE/PubMed article database and by NLM's catalog of book holdings. MeSH is also used by ClinicalTrials.gov registry to classify which diseases are studied by trials registered in ClinicalTrials.gov.
SNOMED-CT
The Systematized Nomenclature of Medicine (SNOMED) is a systematic, computer-processable collection of medical terms, in human and veterinary medicine, to provide codes, terms, synonyms and definitions which cover anatomy, diseases, findings, procedures, microorganisms, substances, etc. It allows a consistent way to index, store, retrieve, and aggregate medical data across specialties and sites of care. Although now international, SNOMED was started in the U.S. by the College of American Pathologists (CAP) in 1973 and revised into the 1990s. In 2002 CAP's SNOMED Reference Terminology (SNOMED RT) was merged with, and expanded by, the National Health Service's Clinical Terms Version 3 (previously known as the Read codes) to produce SNOMED CT.
Text Mining Method
Text mining, also referred to as text data mining, roughly equivalent to text analytics, is the process of deriving high-quality information from text. High-quality information is typically derived through the devising of patterns and trends through means such as statistical pattern learning. Text mining usually involves the process of structuring the input text (usually parsing, along with the addition of some derived linguistic features and the removal of others, and subsequent insertion into a database), deriving patterns within the structured data, and finally evaluation and interpretation of the output. 'High quality' in text mining usually refers to some combination of relevance, novelty, and interestingness. Typical text mining tasks include text categorization, text clustering, concept/entity extraction, production of granular taxonomies, sentiment analysis, document summarization, and entity relation modeling (i.e., learning relations between named entities). Text analysis involves information retrieval, lexical analysis to study word frequency distributions, pattern recognition, tagging/annotation, information extraction, data mining techniques including link and association analysis, visualization, and predictive analytics. The overarching goal is, essentially, to turn text into data for analysis, via application of natural language processing (NLP) and analytical methods. A typical application is to scan a set of documents written in a natural language and either model the document set for predictive classification purposes or populate a database or search index with the information extracted.
Calculation of Phenotype-based Disease Similarity
In the field of information retrieval, text documents or concepts are commonly represented by feature vectors. Here, we describe every disease j by a vector of phenotypes dj

where wi,j quantifies the strength of the association between phenotype i and disease j. The prevalence of the different phenotypes and diseases is very different, for example, there are highly abundant phenotypes like pain, and publication biases towards certain diseases like breast cancer. To account for this heterogeneity, we therefore do not use the absolute co-occurrence Wi,j to measure the strength of an association between phenotype i and disease j, but the term frequency-inverse document frequency60 wi,j:
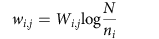
where N denotes the number of all diseases in the dataset and ni the number of diseases where phenotype i appears. Since all phenotypes in our data have at least one associated disease, the potential problem of dividing by zero does not arise.A widely used measure in both text mining and the biomedical literature to quantify the similarity between two concepts is the cosine similarity of the respective vectors. The similarity between the vectors dx and dy of two diseases x and y is calculated as follows:
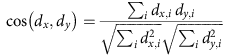
The cosine similarity ranges from 0 (no shared phenotypes) to 1 (identical phenotypes).
Calculation of Gene-based Disease Similarity
To analyze disease relationships, we built a disease co-occurrence matrix based on shared genes between each pair of diseases. We first calculated the uniqueness of each gene i as follows:
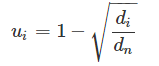
where di is the number of diseases associated with each gene i and dn is the number of diseases in the data set. Note that the fewer number of diseases related to a gene, the higher the possible uniqueness score for that gene.Next, we created an N × N matrix. For each pair of diseases we added the uniqueness score of each shared gene:
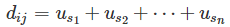
where di,j is a disease pair and usnusn is the uniqueness value for each gene shared between the two. The diagonal elements of the disease co-occurrence matrix, where i = j for di,j , contain the sum of the uniqueness values for all genes related to disease di .