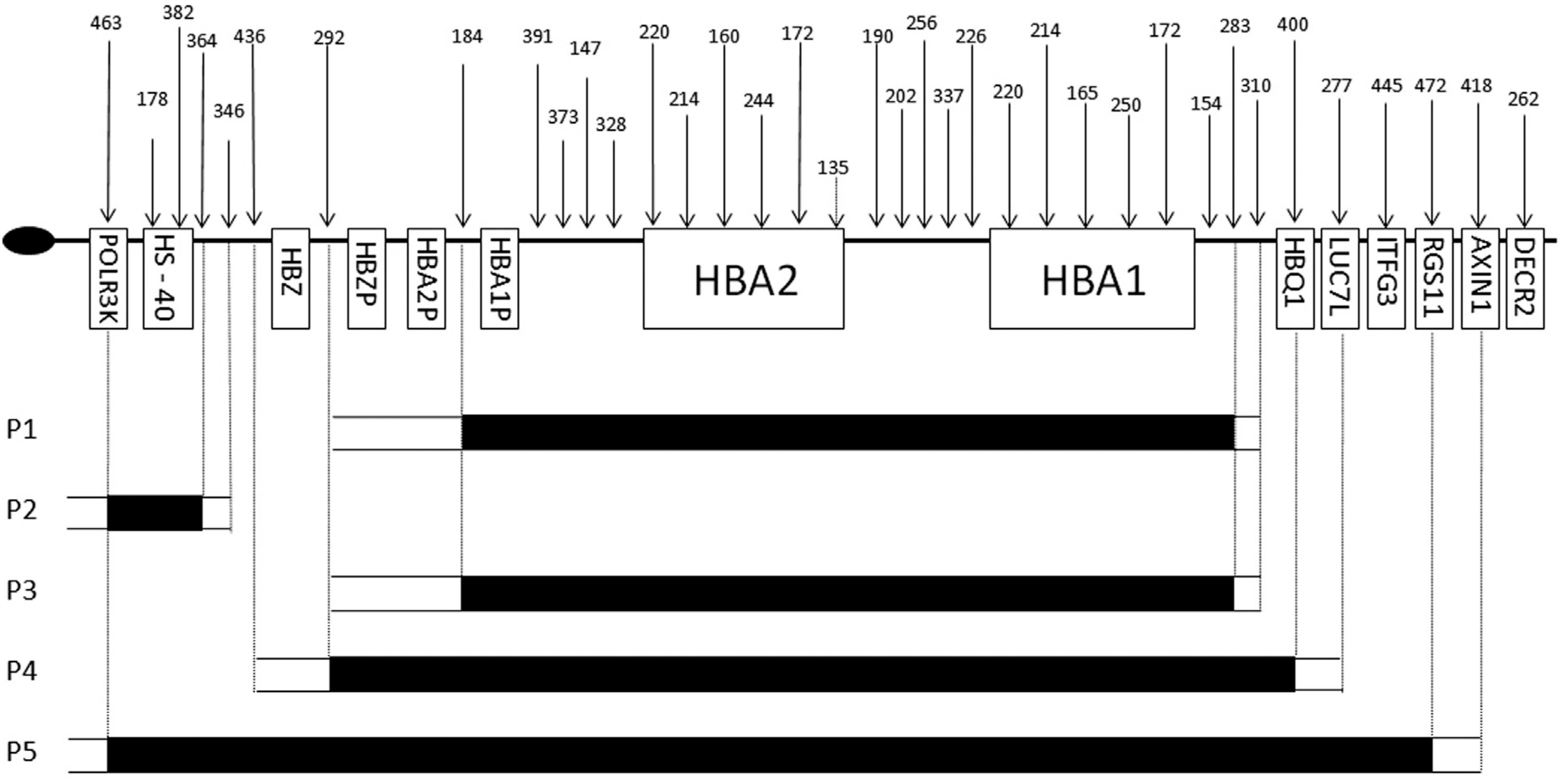
Alpha-thalassemias are among the most common genetic diseases in the world. They are characterized by hypochromic and microcytic anemia and great clinical variability, ranging from a practically asymptomatic phenotype to severe anemia, which can lead to intrauterine or early neonatal death. In Oct 2, 2017, I. Natália O. Mota and others published an article in << Genetics and Molecular Biology >> which title is “Rare α0-thalassemia deletions detected by MLPA in five unrelated Brazilian patients”. Multiplex ligation-dependent probe amplification (MLPA) can be used to detect rearrangements that cause α-thalassemia, particularly large deletions involving the whole α cluster and/or deletions in the HS-40 region. Here, MLPA was used to investigate the molecular basis of α-thalassemia in five unrelated patients, three of whom had Hb H disease. There results illustrate the diversity of α-thalassemia deletions in the Brazilian population and highlight the importance of molecular investigation in cases that present with microcytosis and hypochromia without iron deficiency and normal or reduced Hb A2 levels.
Read More