1. Introduction
X-CNV is an analysis platform to predict CNV pathogenicity using an XGBoost classifier, which calculates a meta-voting prediction (MVP) score to quantitatively evaluate disease-causing probability. It consists of the most comprehensive CNV data and annotations by integrating various publicly available genetic variant repositories. We took into account a set of informative features covering the genomics, genome region, variation types, and population genetics properties to boost the prediction power. More importantly, a meta-voting prediction (MVP) score was proposed to measure the CNV pathogenic effect quantitatively.
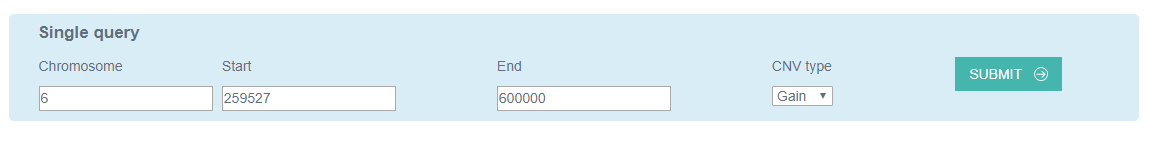
2. Single query
The single query module allows users to provide the chromosome, start, end, and CNV type and submit to the analysis interface. The analysis interface accepts chromosomes including autosomes (1, 2, …, and 22) and sex chromosomes (X and Y). The start and end positions should be integers and the former must be less than the latter.
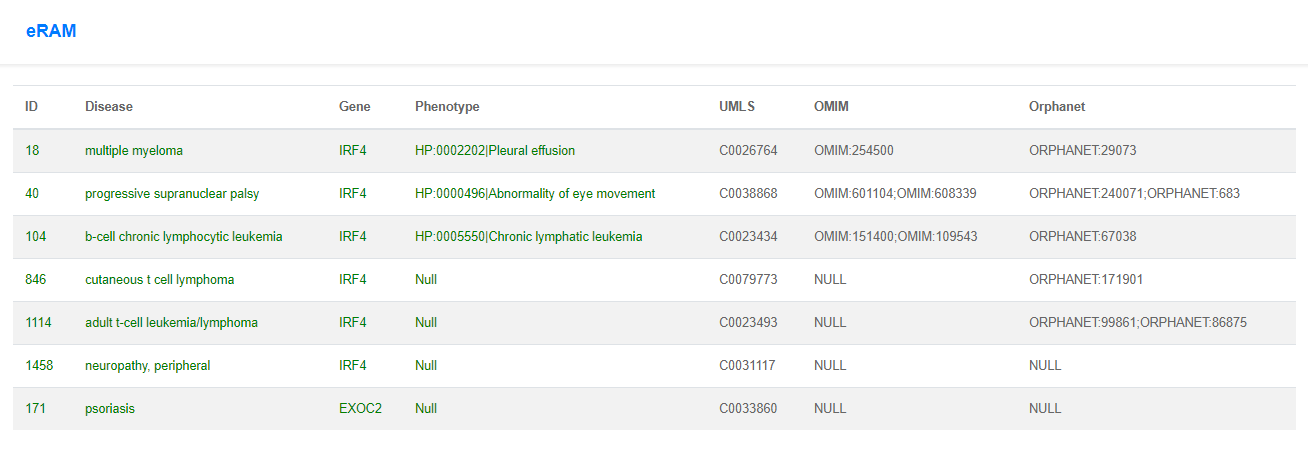
When the data is submitted, the current page will jump to another website, which presents the basic information (including CNV length, MVP score, and genes located within the CNV region), features, and annotations from databases including eRAM, DGV, and GTEx. The CNVs in DGV overlapping with the CNV region provided by the users will be visualized by IGV plugin.
Basic information:

Features:

Annotations:

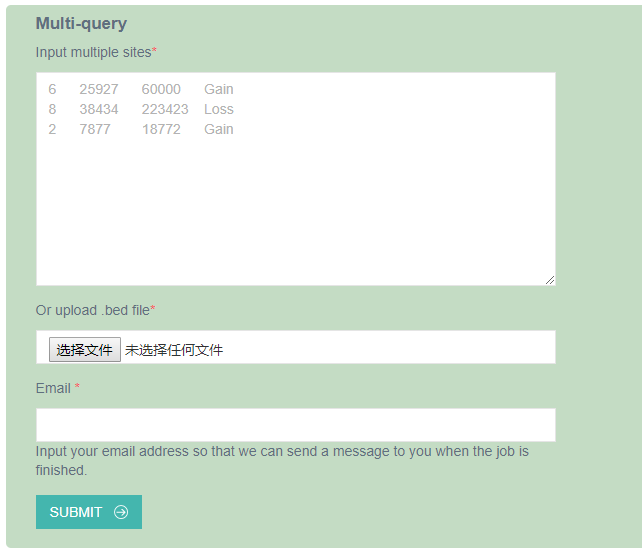
3. Multi-query
The multi-query module allows users to upload the chromosome, start, end, and CNV type of multiple CNVs (Tab-separated values are required), or a BED format file to the analysis interface. The analysis interface accepts chromosomes including autosomes (1, 2, …, and 22) and sex chromosomes (X and Y). The start and end positions should be integers and the former must be less than the latter.
4. Local version
The local version of X-CNV model can be downloaded from the link. Users can follow the instructions of the download page.